A hospital procures new clinical monitors based on price and picture quality, only to face an audit failure months later. The devices lack the required regulatory certification, creating significant legal and operational risk.
CE MDR certification elevates medical monitors from simple electronics to regulated devices with full lifecycle traceability. For hospitals, making it a procurement requirement is essential for ensuring clinical safety, legal compliance, and supply chain stability.

When equipping a hospital with essential technology, the procurement process must go beyond technical specifications and price points. For medical monitors used in clinical settings, regulatory compliance is not just a formality—it is a fundamental prerequisite for patient safety and institutional integrity. The European Union’s Medical Device Regulation (MDR 2017/745)1 has fundamentally changed the landscape, reclassifying clinical displays as regulated medical devices. This shift imposes stringent requirements on manufacturers, covering everything from design and risk management to post-market surveillance. For hospitals, understanding and mandating CE MDR certification is no longer optional. It is the only way to ensure that the displays you purchase are safe, effective, and legally placed on the market. This article will demystify the CE MDR framework, outline the risks of non-compliance, and provide a practical guide for building an MDR-first procurement strategy that protects your patients, your clinicians, and your institution.
What are CE and MDR certification?
CE marking is the EU market-access label. For medical monitors, “CE under MDR (EU 2017/745)” proves the device passed medical-grade conformity assessment with lifecycle controls, not just electronics rules like LVD/EMC/RoHS.
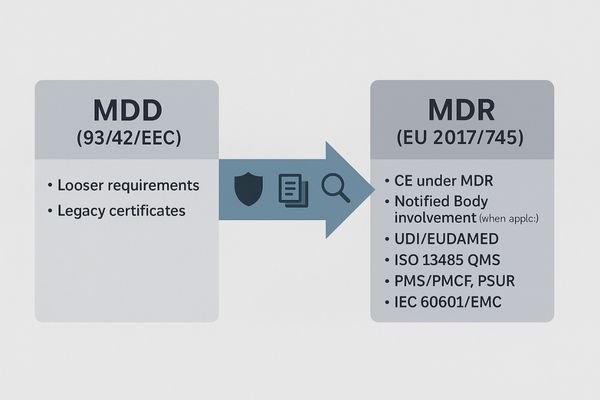
- CE (CE Marking): An EU/EEA market-access mark indicating conformity with EU legislation. Consumer electronics often rely on LVD/EMC/RoHS, but that alone is not sufficient for medical devices.
- MDR (EU 2017/745): The medical device “mother law” covering risk classification, conformity assessment (with Notified Body where applicable), technical documentation, clinical evaluation/PMCF, QMS (ISO 13485), UDI with EUDAMED registration, PMS/PSUR, labeling and IFU.
- How they relate: A medical monitor must comply with MDR first, then affix the CE mark—hence “CE under MDR” (different from “CE under LVD/EMC” for general electronics).
- Applied to medical/surgical monitors (Intended Use): If the manufacturer declares clinical viewing/diagnosis/surgical use, the display becomes a medical device (or accessory). Class depends on intended use (e.g., Class I or IIa), and the route may require Notified Body involvement.
- Typical compliant artifacts you should see: CE + UDI on device/packaging; Declaration of Conformity (DoC) citing MDR and applicable harmonized standards; manufacturer/authorized representative details and EUDAMED entries; ISO 13485 evidence; CER/PMCF, ISO 14971 risk file, PMS procedure; multilingual IFU.
Buyer’s 5 quick checks:
- DoC scope: States EU 2017/745, not only LVD/EMC.
- Risk class & NB involvement: Clear pathway; certificates verifiable.
- UDI labeling: On device/pack; vendor can provide UDI-DI list.
- EUDAMED records: Manufacturer/SRN and device entries searchable and active.
- QMS & clinical evidence: ISO 13485 certificate; CER/PMCF/PSUR summaries or controlled access available.
In short: a hospital-grade monitor in the EU = MDR conformity assessment + CE marking + UDI + ongoing PMS; electronics-only CE is not sufficient for clinical procurement.
What CE MDR actually requires
MDR compliance is not a one-time test. It is a commitment to a continuous cycle of evaluation, documentation, and surveillance that lasts for the entire product lifetime.
MDR elevates monitors from "general electronics" to regulated devices, demanding lifecycle controls including clinical evaluation, risk management, usability engineering, comprehensive safety testing, UDI traceability, and continuous post-market surveillance.
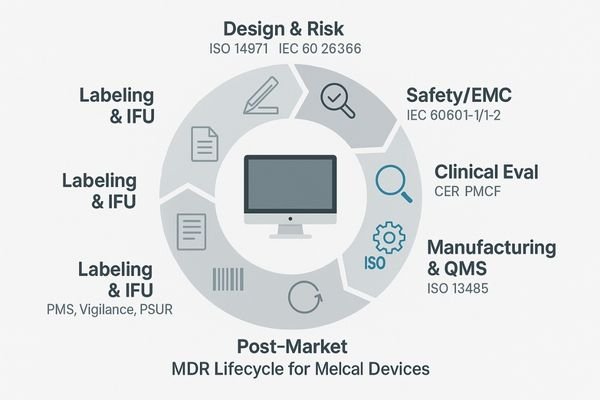
The core requirement of CE MDR is the shift in perspective: a medical monitor is no longer treated as a piece of general-purpose IT hardware but as a purpose-built medical device subject to the same level of control as other clinical equipment. This imposes a holistic, lifecycle-based regulatory burden on the manufacturer. It begins with the Clinical Evaluation Report (CER), where the manufacturer must systematically gather and analyze clinical data to prove that the device is safe and performs as intended for its stated clinical use. This is not just a technical test but a validation of the product’s clinical utility.
From Design to Disposal
This is supported by a comprehensive risk management file (ISO 14971) that identifies, evaluates, and mitigates all potential hazards associated with the device’s use. Usability engineering (IEC 62366) ensures the device interface is safe and intuitive for clinicians. Mandatory compliance with IEC 60601-1 (electrical safety) and 60601-1-2 (EMC) guarantees the device can operate safely in a hospital’s complex electronic environment. The Unique Device Identification (UDI)2 system provides cradle-to-grave traceability, with each device registered in the central EUDAMED database. This system is fed by the manufacturer’s Post-Market Surveillance (PMS) plan, which mandates proactive data collection to monitor the device’s real-world performance and safety long after the sale.
Clinical, legal, and supply risks without MDR
Procuring non-MDR monitors might seem like a cost-saving measure, but it introduces a cascade of hidden risks that can prove far more expensive in the long run.
Non-MDR monitors create risks of inconsistent image quality and safety gaps. Hospitals face audit failures, insurance disputes, product quarantines during an incident, and costly fleet-wide replacements.

Choosing a monitor without valid CE MDR certification exposes a hospital to a triad of critical risks: clinical, legal, and supply chain.
Clinical and Legal Dangers
Clinically, a non-MDR device comes with no guarantee of performance or safety consistency. Without a CER or adherence to standards like DICOM Part 14 (GSDF tracking), image fidelity can be unreliable, potentially masking or mimicking pathology. Labeling and Instructions For Use (IFU) may be unclear or incomplete, and there is no mandated vigilance system for reporting adverse incidents. Legally, the risks are severe. A hospital knowingly procuring and using non-compliant medical devices3 can face failures in regulatory audits, potentially leading to fines and sanctions. In the event of a misdiagnosis linked to the display, an institution may face insurance coverage disputes and significant medico-legal liability, as it cannot prove it followed due diligence in its procurement process.
Supply Chain and Operational Disruption
From a supply chain perspective, non-compliant devices are a ticking time bomb. If a regulatory authority discovers the non-compliance, they can order the entire fleet of devices to be quarantined or recalled, causing massive operational disruption. This forces the hospital into a costly and unplanned emergency replacement cycle. It also creates a swap-out risk mid-lifecycle, where a supplier discontinues a non-compliant product, leaving the hospital with no source for identical replacements or spare parts, destroying fleet standardization.
How MDR shapes your specification and RFP
MDR provides a powerful framework for writing clear, enforceable, and evidence-based specifications for your Request for Proposal (RFP), moving beyond subjective marketing claims.
Translate MDR requirements into concrete RFP specifications: demand a declared intended use, a CER to back clinical claims, GSDF/luminance data, safety reports, a valid UDI, and a documented PMS plan.

The MDR framework provides a clear blueprint for strengthening your procurement documents. Instead of using vague terms like "high-quality medical display," your Request for Proposal (RFP) can and should demand specific, verifiable evidence tied directly to MDR requirements. Start by requiring the vendor to provide the device’s official "Intended Use" statement, as this legally defines what the monitor is designed and approved for. All clinical performance claims, such as "improved lesion detection," must be substantiated with a summary of the Clinical Evaluation Report (CER).
Building an Evidence-Based Tender
Technical specifications should be tied to performance standards. Require documented proof of DICOM Part 14 GSDF compliance4, along with factory test data for luminance stability over time (ΔL/L) and uniformity limits. Specify acceptance and maintenance thresholds once to guide vendors and your QA team: GSDF error within ±5%, uniformity within 15%, and luminance stability ΔL/L within 10%. Mandate the submission of test reports or certificates for IEC 60601-1 (electrical safety) and IEC 60601-1-2 (EMC) to ensure the device is safe for the hospital environment. Crucially, your RFP must require the vendor to provide the device’s UDI-DI code and the manufacturer’s Single Registration Number (SRN) from the EUDAMED database. Finally, request an overview of the manufacturer’s Post-Market Surveillance (PMS) cadence and confirm that the Instructions For Use (IFU) are available in all required local languages.
Verification steps buyers should follow
Trust but verify. A vendor’s claims of MDR compliance must be checked against objective evidence through a systematic verification process.
Request the Declaration of Conformity and CE certificate, verify the UDI in EUDAMED, review the CER summary and safety reports, confirm factory ISO 13485, and perform physical acceptance tests.

Once you receive responses to your RFP, a structured verification process is essential to validate a vendor’s claims. Do not rely on marketing brochures alone.
- Declaration of Conformity and CE certificate: confirm the Notified Body number, certificate validity dates, and that the model family is explicitly covered by scope.
- EUDAMED verification: check the device’s UDI-DI and the manufacturer’s SRN are present and active in the public database.
- Clinical Evaluation and risk file: request a CER summary and verify alignment with intended use and labeling; confirm ISO 14971 risk controls are implemented.
- Safety/EMC evidence: collect IEC 60601-1 and 60601-1-2 third-party test report numbers and issuing bodies.
- Factory quality system: verify an ISO 13485 certificate number and covered site addresses.
- IFU and labeling: confirm languages required by your hospital are available and labels include UDI, lot/serial, and warnings per MDR.
Quantifying the Degradation
Build physical acceptance testing into your process for sample units and first-article deliveries. Measure luminance uniformity across multiple points, evaluate luminance stability over a warm-up and duty cycle, confirm GSDF tracking using acceptance patterns, validate latency for clinical video paths if applicable, and record results against thresholds: GSDF error within ±5%, uniformity within 15%, luminance stability ΔL/L within 10%. Define sampling ratios and periodic re-tests in your QA plan (for example, at delivery and quarterly or semi-annually).
Solution: build an MDR-first vendor checklist
To ensure consistency and rigor, standardize your evaluation process across all departments with a weighted, evidence-based vendor checklist focused on MDR compliance.
Create a standardized MDR checklist covering regulatory documents, technical benchmarks, post-market obligations, and service level agreements. Score vendors on the evidence they provide, not on their promises.
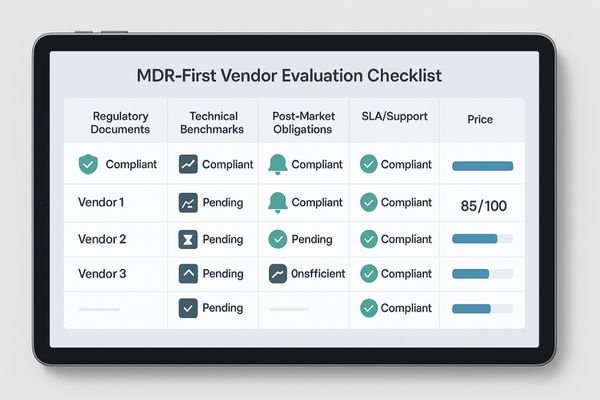
The most effective way to implement an MDR-first procurement strategy5 is to create a standardized vendor checklist that can be used by clinical, technical, and purchasing departments. This moves the evaluation from a subjective discussion to an objective, data-driven process. The checklist should be structured around key MDR pillars and can be weighted based on importance. The first section should cover the complete set of regulatory documents: the DoC, CE certificate, ISO 13485 certificate, and proof of EUDAMED registration. The second section should focus on technical benchmarks, requiring vendors to provide verifiable factory data for DICOM/GSDF compliance, luminance stability over time (ΔL/L), and screen uniformity. The third section must address post-market obligations, clarifying the vendor’s PMS/PMCF plan and their policy for notifying customers of reportable incidents or corrective actions. A fourth section should detail the service level agreement (SLA), including policies on spare parts availability, firmware updates, and warranty support over the device’s expected lifetime. By scoring each vendor against this evidence-based checklist, you can make a defensible decision based on documented compliance and calculated risk, not just marketing claims.
Recommended MDR-Compliant Monitors
| Model | Primary Use Case | Key MDR-Aligned Features |
|---|---|---|
| MD32C | 3MP Diagnostic Color | DoC/CE scope, ISO 13485 factory, DICOM GSDF data, ΔL/L and uniformity logs, UDI/EUDAMED |
| MD52G | 5MP Diagnostic Grayscale | DoC/CE scope, GSDF acceptance ready, luminance stability records, IEC 60601 reports |
| MD22CA | 2MP Clinical Review | Standardized presets, QA logging readiness, UDI labeling and IFU languages |
| MS321PB | 32″ 4K Surgical | Declared intended use for clinical viewing, low-latency I/O, 60601/EMC reports, EDID policy |
| MS550P | 55″ OR Wall Display | 4K quad-view for teams, documented integration policy, labeling and UDI traceability |
With a standardized, evidence-first checklist in place, procurement decisions translate directly into lower risk and smoother audits.
Business value: risk control and total cost
Adopting an MDR-first procurement strategy is not about adding bureaucracy. It is a strategic decision that delivers tangible business value by controlling risk and optimizing costs.
MDR compliance reduces audit risk, speeds up tender approvals, and cuts downtime with predictable QA. It extends the device lifecycle through controlled updates, lowering the total cost of ownership.

Prioritizing CE MDR certification in procurement delivers significant and measurable business value far beyond simple compliance. The most immediate return is in risk control. By mandating MDR, you drastically reduce the risk of failing a regulatory audit and protect the institution from the legal and financial fallout of using non-compliant equipment. This robust due diligence also simplifies and speeds up the internal tender approval process, as the regulatory requirements are clear and non-negotiable. Operationally, MDR-compliant devices, with their emphasis on stable and predictable performance, reduce clinical downtime. Their quality and consistency can be managed through a predictable QA program rather than reactive, emergency service calls. This reliability, combined with a manufacturer’s commitment to post-market support and controlled firmware updates, extends the effective clinical lifecycle of the monitors, delaying capital expenditure. Together, these factors significantly lower the total cost of ownership (TCO) while simultaneously safeguarding clinical outcomes and patient safety.
FAQ: practical procurement questions
As procurement teams begin to integrate MDR requirements into their workflow, several practical questions about implementation and scope consistently emerge.
Yes, MDR is likely needed for any display used for clinical viewing. Compare vendors with a weighted matrix, and only accept legacy MDD stock with a clear, documented transition plan.
Do we need MDR for non-diagnostic displays?
If a display is used for any form of clinical viewing, decision-making, or data recording—even for tasks like reviewing patient charts at a nurses’ station or displaying vitals in a patient room—it is very likely to be considered a medical device under the MDR’s broad definition. The key is the intended use. If the manufacturer intends for it to be used in a medical environment for clinical purposes, or if the hospital implements it for such a purpose, it should be treated as a medical device requiring MDR compliance.
How can we compare vendors quickly and fairly?
The most effective method is to use a weighted scoring matrix based on your MDR-first checklist. Assign points to each requirement: full and current documentation, verifiable performance metrics, a clear PMS and service plan, and price. This forces a data-driven comparison and prevents price from being the sole determining factor when compliance and safety are at stake.
Can we still accept legacy stock compliant with the old MDD?
The MDR includes transitional provisions, but they are complex and time-limited. Accepting a device that is only compliant with the previous Medical Device Directive (MDD) is risky. You should only consider it if the vendor can provide clear, official documentation from their Notified Body outlining the specific transitional arrangements for that device, including the exact date by which it must be fully MDR-compliant or taken off the market. Insist on a clear plan for future support and replacement.
Conclusion
CE MDR transforms monitor procurement from a simple purchase into a verifiable system, compelling you to select vendors who manage the full product lifecycle, not just ship a screen.
- Exploring the key changes in MDR 2017/745 will help you grasp the new compliance landscape for medical devices. ↩
- Exploring UDI helps grasp its importance in traceability and safety monitoring of medical devices throughout their lifecycle. ↩
- Understanding the risks associated with non-compliant medical devices can help healthcare professionals make informed decisions and ensure patient safety. ↩
- Exploring DICOM Part 14 GSDF compliance will help you grasp its significance in ensuring medical imaging quality and safety. ↩
- Explore this link to understand the principles and benefits of an MDR-first procurement strategy for better vendor selection. ↩