CE marking for medical grade monitors depends on intended purpose, claims, and regulatory pathway—not the “medical grade” label. The fastest path is to build a traceable claim-to-evidence package: define scope, freeze a baseline configuration, map requirements to tests, and keep labeling/IFU and post-market controls consistent with verified boundaries.
Core CE documents typically include: device description and variants, a risk management file, a requirements-to-evidence matrix, safety/EMC and performance verification reports, controlled labeling/IFU, and PMS/PMCF/vigilance plans. Delays most often come from inconsistent claims across marketing, labeling, and evidence, or missing traceability from requirements to test reports.
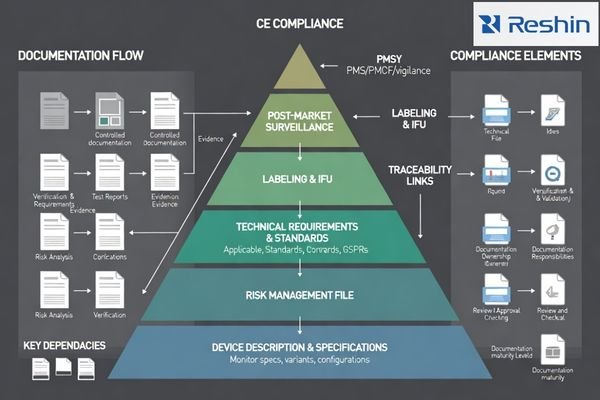
A strong submission treats documentation as one integrated system: every claim must trace to a controlled configuration, a risk control1, and verification evidence with acceptance criteria. When that “spine” is built early, the package is easier to assess, easier to maintain after launch, and less likely to break during updates or field replacements.
What is the scope of CE marking for medical grade monitors?
CE marking scope for medical grade monitors depends on intended clinical purpose and regulatory classification within EU medical device frameworks.
CE marking starts by defining intended purpose, device role (device/accessory/component), and regulatory pathway, because the required evidence depth depends on your claims and use environment. Monitors positioned for diagnostic interpretation or clinical visualization typically need structured technical documentation demonstrating applicable GSPR compliance, supported by risk management and verification evidence that matches the stated conditions of use.
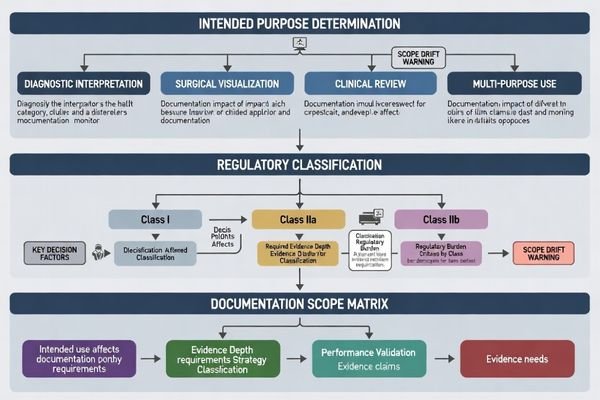
Scope definition is a traceability decision: your intended purpose drives which standards and evidence types are relevant, while your labeling/IFU must stay inside verified performance boundaries. A common delay driver is scope drift—marketing or sales claims expanding beyond what the technical file actually supports.
Clinical Use Definition
Clarifying whether monitors support diagnostic reading, surgical visualization, or non-diagnostic review affects performance expectations, clinical evaluation strategy, and evidence depth needed to support claims, particularly around image quality stability, environmental conditions, and lifecycle controls.
Regulatory Pathway Selection
The regulatory pathway2 determines documentation requirements based on whether monitors are marketed for diagnostic interpretation requiring structured clinical evaluation or for surgical support requiring focused performance validation under specified operating conditions.
Which technical documentation elements are always expected for CE marking?
Core technical documentation elements establish traceability from regulatory claims to supporting evidence across all medical grade monitor applications.
Across most CE pathways, assessors expect a traceable set: device description/specs, intended use, variants/configurations, and a risk management file, plus a requirements-to-evidence matrix linking GSPR/standards clauses to test reports and design outputs. Typical evidence includes electrical safety and EMC reports, relevant mechanical/environmental justifications, software/firmware lifecycle documentation when applicable, and verification of essential performance tied to the claimed clinical workflow.
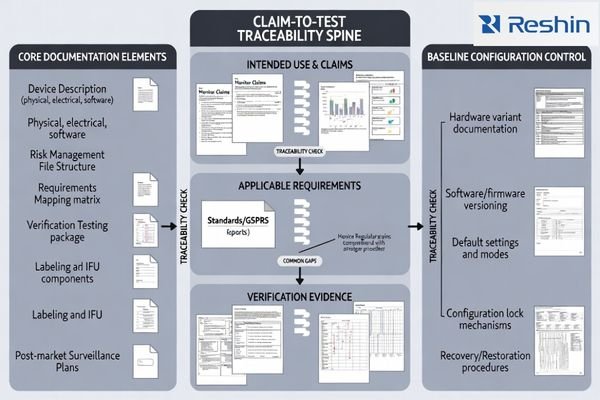
A practical rule is: if you can’t reproduce the baseline configuration3, you can’t defend the claim. Build the “claim-to-test” spine early so every label/IFU statement is backed by a method, acceptance criteria, and a controlled configuration that can be reproduced in production and service.
Evidence packages commonly include electrical safety and EMC test reports, biocompatibility justification where user contact is relevant, mechanical and environmental assessments covering cleaning and mounting expectations, software and firmware lifecycle documentation where embedded software influences safety or performance, and verification of essential performance characteristics tied to clinical workflows.
What risk management and clinical evaluation documents are critical?
Risk management and clinical evaluation documents connect safety and essential performance to real clinical use conditions.
Risk management is central for monitors because use errors and hidden changes can affect essential performance in clinical environments. A complete risk file identifies hazards (e.g., mode drift, misconfiguration, cleaning/ingress, mounting stability, EMC susceptibility, and software/firmware change effects), defines controls (design, labeling, processes), and verifies effectiveness. Clinical evaluation aligns evidence to claims, bounded by clear conditions of use.
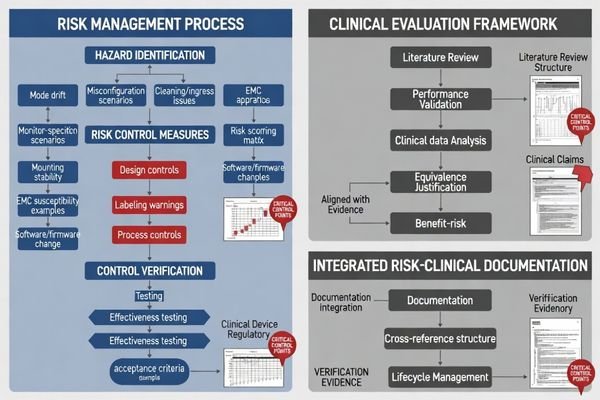
The monitor-specific risk focus should be practical and testable: settings that can change displayed output, stability over time and across state changes, and service/update actions that could silently alter behavior. The most common submission weakness is listing hazards without proving that the chosen controls are effective in the intended configuration.
| Risk Category | Typical Hazards | Control Methods | Verification Requirements | Documentation Elements |
|---|---|---|---|---|
| Electrical Safety | Shock, thermal burns | Isolation, protection circuits | Safety standard testing | Test reports, design rationale |
| EMC Susceptibility4 | Signal interference | Shielding, filtering | EMC compliance testing | Performance under interference |
| Mechanical Stability | Mounting failure | Design limits, warnings | Load testing, instructions | Mounting guidance, limitations |
| Configuration Control | Clinical mode drift | Locked settings, indicators | Mode stability testing | Picture mode validation |
| Software Behavior | Uncontrolled changes | Version control, validation | Software lifecycle | Change control procedures |
Clinical evaluation documentation should demonstrate that image-related performance claims are realistic, repeatable, and bounded by clearly stated conditions of use rather than open-ended marketing language that cannot be supported during conformity assessment or post-market surveillance activities.
Risk management should show how identified hazards are controlled through design features like locks and alarms, labeling including warnings and contraindications, and processes such as factory calibration and field service controls, with verification that controls work under defined operating conditions.
How do labeling, IFU, and post-market plans support CE compliance?
Labeling, instructions for use, and post-market surveillance plans are controlled documents that must maintain consistency with technical files throughout product lifecycles.
Labeling and IFU are controlled compliance documents: they must match the technical file, risk controls, and verified performance boundaries. They should state intended purpose, operating limits, compatible inputs, configuration restrictions, cleaning instructions, mounting guidance, and validated/unsupported modes. Post-market plans (PMS/PMCF where applicable, vigilance) define how field data, complaints, and reportable events are handled to keep compliance maintainable.
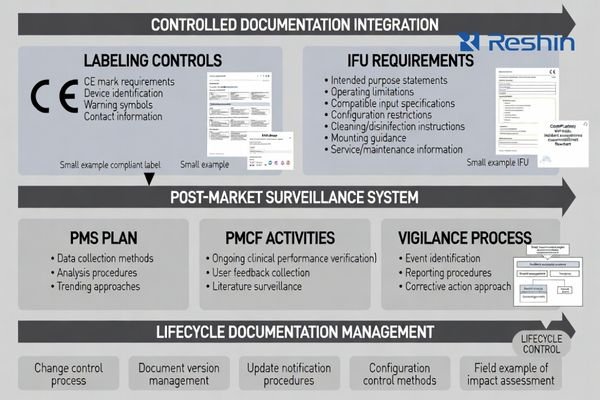
Post-market documentation closes the lifecycle loop through PMS plans defining field feedback collection and analysis, PMCF approaches explaining ongoing clinical performance confirmation, and vigilance processes handling reportable events with clear escalation procedures.
Controlled Labeling Requirements5
Labels and IFU must reflect verified performance boundaries and include warnings about settings that can change perceived output, instructions for maintaining validated configurations, and guidance for service and replacement that protects clinical repeatability across deployment environments.
Post-Market Surveillance Integration
For medical monitors deployed across multiple rooms, strong change-control messaging in IFU and service documentation often determines the difference between maintaining compliance after launch versus experiencing non-compliance after updates or field modifications.
How to build a CE technical documentation package efficiently for medical grade monitors?
Efficient CE documentation development treats documentation as a traceable claim-to-evidence system rather than independent file collections.
Building comprehensive CE technical documentation requires systematic approaches that maintain traceability and consistency.
The fastest way to build a CE-ready package is to lock a baseline configuration, build a requirements mapping spine (claims → clauses → controls → tests), and embed lifecycle controls so updates and replacements don’t break compliance. If claims, labeling, and evidence are consistent, assessment is smoother; if they diverge, delays are likely—even when test reports exist.
Baseline Configuration Control
Start by freezing baseline configurations6 including hardware variants, inputs, firmware, default modes, and lockable settings that tests and labeling will reference, because uncontrolled variants create rework and gaps during assessment processes. Define a “baseline package” field set early (variants, interfaces, firmware, default modes/locks, referenced test reports, and restore steps) so the validated configuration can be reproduced in production and service.
Build requirements mapping that connects intended use and key claims to applicable GSPR and standards clauses, then to risk controls, and finally to specific verification reports with clear acceptance criteria that support regulatory decision-making.
Risk Management Integration
Structure risk management so that highest-likelihood, highest-impact clinical-use hazards including misconfiguration, mode drift, cleaning and ingress issues, mounting stability, EMC susceptibility, and change-control risks are each tied to concrete controls and verification methods. Change control is part of compliance, not an afterthought: assess impact, re-verify affected requirements, update documentation, and ensure the released configuration matches labeling and IFU.
Ensure labeling, IFU, and service documentation match validated boundaries and include lifecycle controls such as update procedures, configuration restore processes, and replacement equivalence guidance so compliance remains stable after launch rather than degrading with updates and field replacements.
FAQ
Are "medical grade" claims enough to apply CE marking as a medical device monitor?
No; CE documentation depends on intended purpose and claims—define the clinical role and build evidence that supports those claims under controlled conditions.
Do we need a full technical file even if the monitor is used only for surgical visualization?
If it is placed on the market as a medical device (or accessory) for that purpose, you still need technical documentation demonstrating applicable safety and performance requirements.
What documents usually take the longest time to prepare?
Risk management with verification evidence, requirements mapping, and consolidated test reports often dominate timelines because they require traceability and controlled configurations.
How should we handle firmware updates without breaking CE compliance?
Use change control: assess impact, re-verify affected requirements, update documentation, and ensure the released configuration matches labeling and IFU.
Do we need clinical trials for a medical monitor?
Often you can rely on literature and performance validation aligned to claims, but the need depends on classification, claims, and equivalence arguments.
What is the most common reason CE assessments get delayed for monitors?
Inconsistent claims across marketing, labeling, and evidence, plus missing traceability from requirements to test reports, are frequent causes of delay.
Conclusion
The core documents for CE marking of medical grade monitors establish traceability from intended use and clinical claims to verified risk controls and performance evidence through systematic documentation approaches. Essential elements include clear device descriptions with controlled variants, comprehensive risk management files, requirements-to-evidence matrices, core safety and EMC verification reports, controlled labeling and instructions for use, and post-market surveillance plans that maintain compliance over product lifecycles. Success depends on understanding that CE marking represents ongoing compliance rather than one-time paperwork exercises.
Our experience at Reshin demonstrates that the fastest route to robust CE submissions involves defining regulatory scope early, building integrated claim-to-test frameworks, and locking baseline configurations that can be reproduced consistently in production and service environments. When technical documentation maintains consistency across marketing claims, risk management, verification evidence, and post-market activities, healthcare organizations can achieve maintainable CE compliance that supports both regulatory approval and long-term market success while protecting clinical performance and safety throughout product lifecycles.
✉️ info@reshinmonitors.com
🌐 https://reshinmonitors.com/
-
Exploring risk control strategies can help you mitigate potential issues in your projects, leading to smoother execution and better outcomes. ↩
-
Exploring regulatory pathways helps clarify documentation requirements and ensures proper compliance for medical device marketing. ↩
-
Understanding baseline configuration is crucial for ensuring reproducibility in tests, which is essential for validating claims. ↩
-
This resource will provide insights into testing and mitigating EMC susceptibility, vital for device performance in various environments. ↩
-
Exploring Controlled Labeling Requirements helps ensure that medical devices meet safety standards and regulatory compliance, which is essential for manufacturers. ↩
-
Understanding baseline configurations is crucial for maintaining control and consistency in product development, ensuring quality and compliance. ↩